Le VENT-02 est une petite molécule puissante, sélective et à pénétrance cérébrale. Actif par voie orale, il est positionné comme le meilleur de sa classe pharmacologique, avec le potentiel de changer le traitement de la maladie de Parkinson en soulageant les symptômes et, de surcroît, en modifiant le cours de la maladie. Après avoir passé l’épreuve d’un essai de phase I mené chez des volontaires sains, le VENT-02 est désormais en cours d’essai clinique de phase II pour le traitement de la maladie de Parkinson.
VENT-02
La protéine NLRP3 appartient à une famille de protéines appelées « inflammasomes ». Composante clé du système immunitaire inné, la NLRP3 joue un rôle important dans de nombreuses maladies inflammatoires, notamment des maladies neurologiques et immunologiques. La NLRP3 est une cible validée sur le plan génétique (par exemple dans les syndromes périodiques associés à la cryopyrine [CAPS]) et sur le plan clinique (ILARISMD [canakinumab], un inhibiteur de l’IL‑1β approuvé pour le traitement de l’arthrite juvénile idiopathique et des CAPS, entre autres).
Le VENT‑02 est en cours d’essais cliniques; suivez le lien pour en savoir plus.
Le VENT-02, un inhibiteur oral de NLRP3 à pénétrance cérébrale qui pourrait être le meilleur de la classe des NLRP3
Les résultats de l’essai de phase I ont montré que le VENT-02 a un index thérapeutique étendu et une pénétrance cérébrale élevée. L’inhibiteur a été bien toléré aux doses cliniques, et aucun signe de toxicité qui limiterait la dose n’a été observé. En outre, le VENT‑02 a exercé une modulation significative des biomarqueurs inflammatoires, montrant notamment une saturation de sa cible par une inhibition de 100 % de l’IL‑1β dans le sang, dans le cadre d’un test de provocation ex vivo mené sur du sang entier.
Biologie de NLRP3
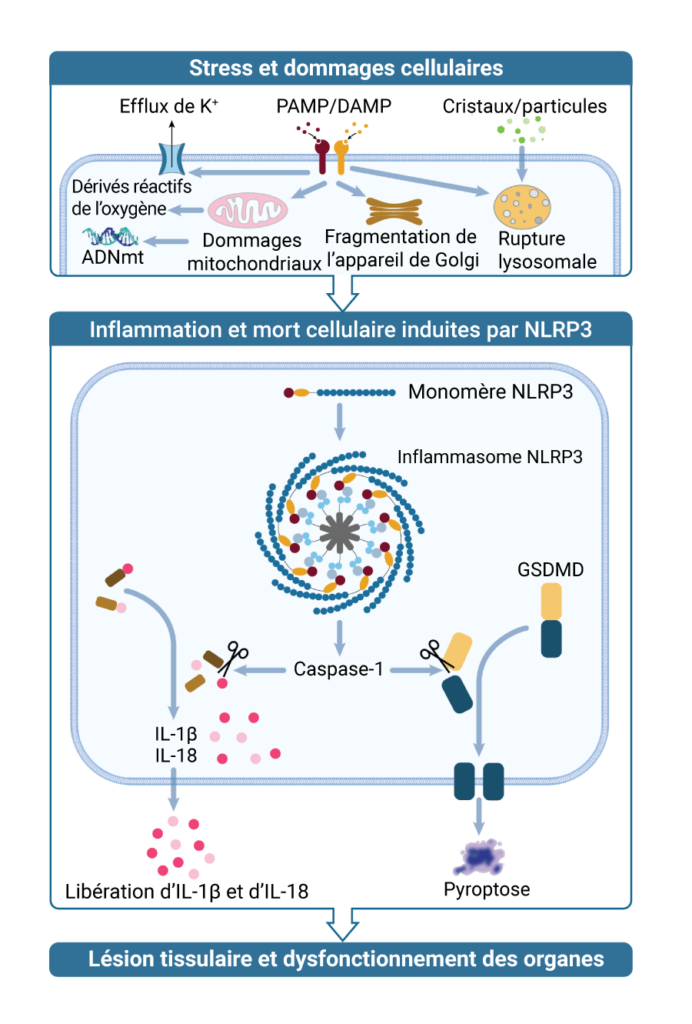
L’inflammasome NLRP3 est un régulateur essentiel du système immunitaire inné qui est activé par des marqueurs de lésions tissulaires, comme les agrégats de protéines anormales découlant du stress cellulaire. Lorsque la protéine NLRP3, sous sa forme monomérique inactive, détecte des dommages à l’intérieur de la cellule, elle se regroupe sous forme d’oligomère, sa conformation active, formant un complexe d’inflammasome hautement organisé. Ce complexe entraîne la libération d’IL‑1β et d’IL‑18 et l’activation de la gasdermine D (GSDMD), qui favorisent en aval une réponse inflammatoire ainsi que la pyroptose, un type de mort cellulaire déclenchée par des signaux pro-inflammatoires. La neuro-inflammation induite par l’activation de NLRP3 perturbe les neurones viables, provoquant des symptômes (p. ex., des troubles cognitifs, des tremblements) et une évolution (mort cellulaire et atrophie cérébrale) des maladies neurologiques classiques (comme la maladie de Parkinson et la maladie d’Alzheimer), et active des mécanismes d’obésité régulés par le système nerveux central (comme l’inflammation dans l’hypothalamus). L’inhibition de l’activation de NLRP3 pourrait moduler cette réponse inflammatoire, et permettre ainsi de traiter les maladies dans lesquelles la protéine NLRP3 est impliquée.
Rôle de l’inhibition de NLRP3 par le VENT-02 dans la maladie de Parkinson (MP)
Le repliement anormal et l’agrégation de l’α-synucléine sont associés au dysfonctionnement et à la dégénérescence des neurones moteurs dopaminergiques dans la MP. Les fibrilles pathogènes de l’α‑synucléine provoquent l’activation de NLRP3 dans la microglie, ce qui entraîne la libération d’IL‑1β, accentuant la formation d’agrégats d’α‑synucléine et leur propagation dans différentes régions du cerveau. Cet ensemencement de l’α-synucléine par NLRP3 a de profondes implications pour le traitement de la MP.
Les traitements actuellement approuvés pour la MP ciblent la diminution des symptômes, et non la perte neuronale sous-jacente, et sont également associés à des événements indésirables et à une issue négative à long terme. De plus, ils n’interviennent pas sur la composante inflammatoire de la MP, qui, selon nous, joue un rôle critique dans la manifestation des symptômes et l’évolution de la maladie.
Le VENT‑02 pourrait changer la donne en matière de traitement, en permettant à la fois de réduire les symptômes et de ralentir l’évolution de la maladie. À court terme, nous nous attendons à une diminution des symptômes moteurs et non moteurs attribuable à une réduction de la réaction inflammatoire induite par NLRP3 en réponse à l’accumulation de l’α‑synucléine. À plus long terme, le VENT‑02 pourrait exercer des effets neuroprotecteurs en prévenant la mort des neurones, avec pour résultat un effet modificateur de la maladie graduel et durable.
Le VENT-01, un inhibiteur oral de NLRP3 à action générale, actuellement en phase I, est le premier candidat au développement sélectionné par Ventus
En septembre 2022, nous avons conclu une entente de licence exclusive avec Novo Nordisk A/S pour développer et commercialiser des candidats-médicaments de notre gamme d’inhibiteurs de NLRP3 à action générale, dont le VENT-01. Pour en savoir plus sur cette collaboration