NLRP3 Biology
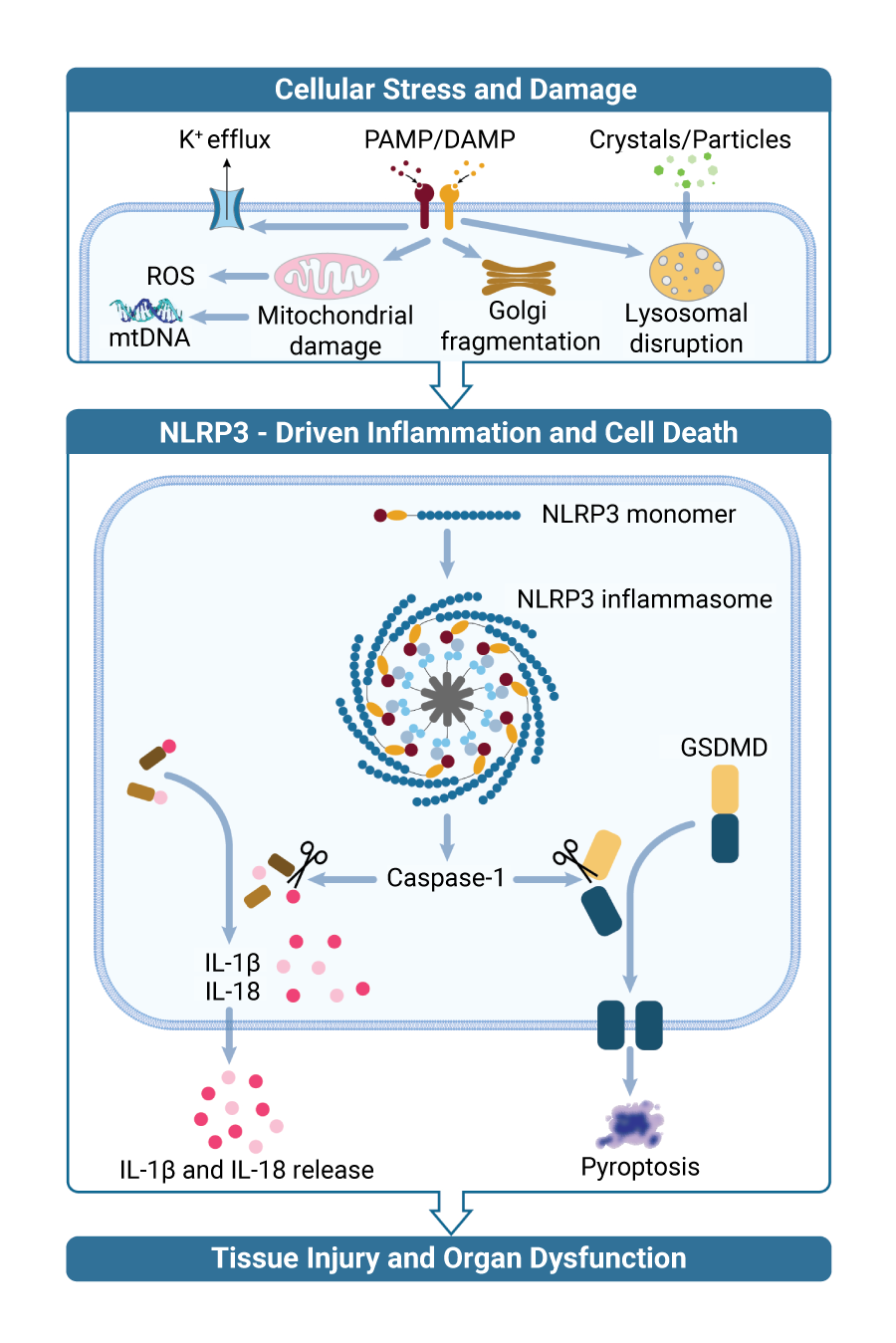
NLRP3 is a critical regulator of the innate immune system and is activated by markers of tissue damage, such as abnormal protein aggregates derived from cellular stress. When inactive monomeric NLRP3 senses intracellular damage, it oligomerizes into its active conformation, forming a highly organized inflammasome complex. This inflammasome complex leads to the secretion of IL-1ß and IL-18, activation of Gasdermin D (GSDMD) to promote a downstream inflammatory response, and pyroptosis, a type of cell death triggered by pro-inflammatory signals. NLRP3-mediated neuroinflammation disrupts viable neurons, causing symptoms (e.g. cognitive impairment, tremors) and disease progression (cell death and brain atrophy) in conventional neurological diseases (such as Parkinson’s and Alzheimer’s) as well as CNS-mediated mechanisms of obesity (such as inflammation in the hypothalamus). Inhibition of NLRP3 activation has the potential to modulate this inflammatory response, resulting in therapeutic potential against NLRP3-mediated diseases.
Role of NLRP3 inhibition with VENT-02 in Parkinson’s Disease (PD)
Misfolding and aggregation of α-synuclein is associated with the dysfunctionality and degeneration of dopaminergic motor neurons in PD. Pathogenic α-synuclein fibrils induce NLRP3 activation in microglia, leading to the release of IL-1β and resulting in further widespread and progressive formation of α-synuclein aggregates in different areas of the brain. This seeding of α-synuclein by NLRP3 has profound therapeutic implications in PD.
Currently approved treatments for PD target symptomatic improvements but not the underlying neuronal loss and are also associated with adverse events and long-term negative outcomes. In addition, they do not address the inflammatory component of PD, which we believe is critical in disease symptoms and progression.
VENT-02 enables a uniquely therapeutic paradigm: improving symptoms as well as slowing disease progression. In the short-term, we anticipate an improvement in motor and non-motor symptoms because of the reduction in NLRP3-driven inflammatory response to a-synuclein accumulation. Over the longer term, VENT-02 may exert neuroprotective effects by preventing neuronal death, resulting in a long-lasting incremental disease-modifying effect.
VENT-01, an oral systemic inhibitor of NLRP3 currently in Phase 1, was the first development candidate nominated by Ventus
In September 2022, we entered into an exclusive development and license agreement with Novo Nordisk A/S to develop and commercialize candidates from our portfolio of systemic NLRP3 inhibitors, including VENT-01. Learn more about the collaboration here.
A Phase 1 clinical trial of VENT-01 is ongoing. Learn more about the Phase 1 study here.